
发布时间:2022-07-25作者:何永芳、邓君点击: 次
还记得今年初,登上热搜的医保药品谈判“灵魂砍价”吗?谈判中,治疗罕见病脊髓性肌萎缩症(SMA)的药物诺西那生钠谈判进行了一个半小时,过程异常艰难。最终,药品由每支70多万元下降为3万多元,新医保目录开启了“小群体”患者的新生。

12岁的来自贺州的脊髓性肌萎缩症(SMA)患儿冬冬(化名)就是受益者之一。日前,冬冬已在桂林市妇幼保健院儿科成功完成了诺西那生钠注射液的首次负荷剂量的鞘内注射,成为桂北地区接受该药治疗的首位患儿。

“每一个小群体都不应该被放弃”2021年12月国家医保局谈判代表张劲妮在国家医保目录药品谈判现场曾说过这样一句话,为了让这句话在桂北地区也变成温暖的现实,让SMA患儿能够在桂林本地就能接受治疗,桂林市医保局对桂林市妇幼保健院提出的罕见病脊髓性肌萎缩症治疗医保费用的支出方式进行了专题研究,给予了专项政策,大大减轻了患者的经济负担,同时也推动了桂北地区罕见病治疗能力的提升。
12岁的冬冬是一个品学兼优的好孩子,在常人看来,这位正值朝气蓬勃年纪的孩子也许会注定有不平凡的人生。“从1岁2个月学走路开始,我们就发现孩子易跌跤、走不远,随着年龄的增长,行走总是比同龄儿困难,不能跑,躯体不同程度的畸形。受限于当时疾病诊断水平,一直未能确诊”。冬冬的妈妈孙女士(化名)回忆道,家人四处求医多年,最终在2021年经基因检测确诊为SMA。但是面临高额的药物治疗费用,这个不幸的家庭并没有觉得柳暗花明。直到2021年SMA特效药——诺西那生钠注射液纳入国家医保,2022年1月1日起正式执行。这也就意味着,SMA患儿将进入有药可用、且用得起的时代。

桂林市妇幼保健院儿科一病区兼儿童重症监护室王菲主任介绍,脊髓性肌萎缩症(SMA)是一种常染色体隐性遗传神经变性病,是导致两岁以下婴幼儿死亡的头号遗传性疾病,目前国内患病人数在3-5万,发病率约为万分之一,是名副其实的罕见病。该疾病可导致严重的、进行性肌肉萎缩和肌肉无力。最终,SMA患者可能丧失行走能力,并且难以完成呼吸和吞咽等基本生活功能。如果不进行治疗,大多数患有最严重疾病类型的婴儿在没有呼吸干预的情况下,可能危及生命。一直以来,全世界对于SMA的治疗措施仅限于呼吸支持、营养支持、骨科矫形等辅助治疗方法,没有针对性的药物可以使用。
2019年,全球首个SMA精准靶向治疗药物“诺西那生钠注射液”在中国获批上市。但是面对近70万一针的药费,大部分SMA家庭完全无法承担,只能望尘莫及。孙女士表示,即使是有援助计划的补贴,一年的药费也不是他们这样的普通家庭能够承担的。终于,他们在新闻中看到了那场“灵魂砍价”。
孙女士告诉笔者,冬冬最大的梦想就是能像小伙伴那样自由行走。
冬冬和家人满怀希望来到桂林市妇幼保健院。“这真的是这类疾病患儿的福音。有的家长告诉我,得知这个好消息后,他们非常激动,甚至抱头痛哭。”负责具体治疗的桂林市妇幼保健院儿童神经专科唐华利副主任医师说,“SMA的治疗就是在与时间赛跑。患者越早诊断、越早开始有效治疗,效果越好。SMA需要终身用药,治疗首年需要注射6针,此后每年需要注射3针。”像冬冬这样的孩子现在开始接受药物、康复等系统治疗,不仅能延缓病程,还能逐渐恢复运动能力。

经前期病情评估,冬冬因为常年发病导致严重的脊柱侧弯,椎间隙狭窄,穿刺给药难度较大。我院儿科汇同麻醉科、B超室、放射科、营养科、康复科、骨科建立了多学科治疗小组(MDT),为冬冬制定了合理有效药物治疗方案,在保障药物治疗的同时也为冬冬的持续肢体运动功能的评估、持续康复、营养管理和心理辅导等后续治疗制定了整体的方案。
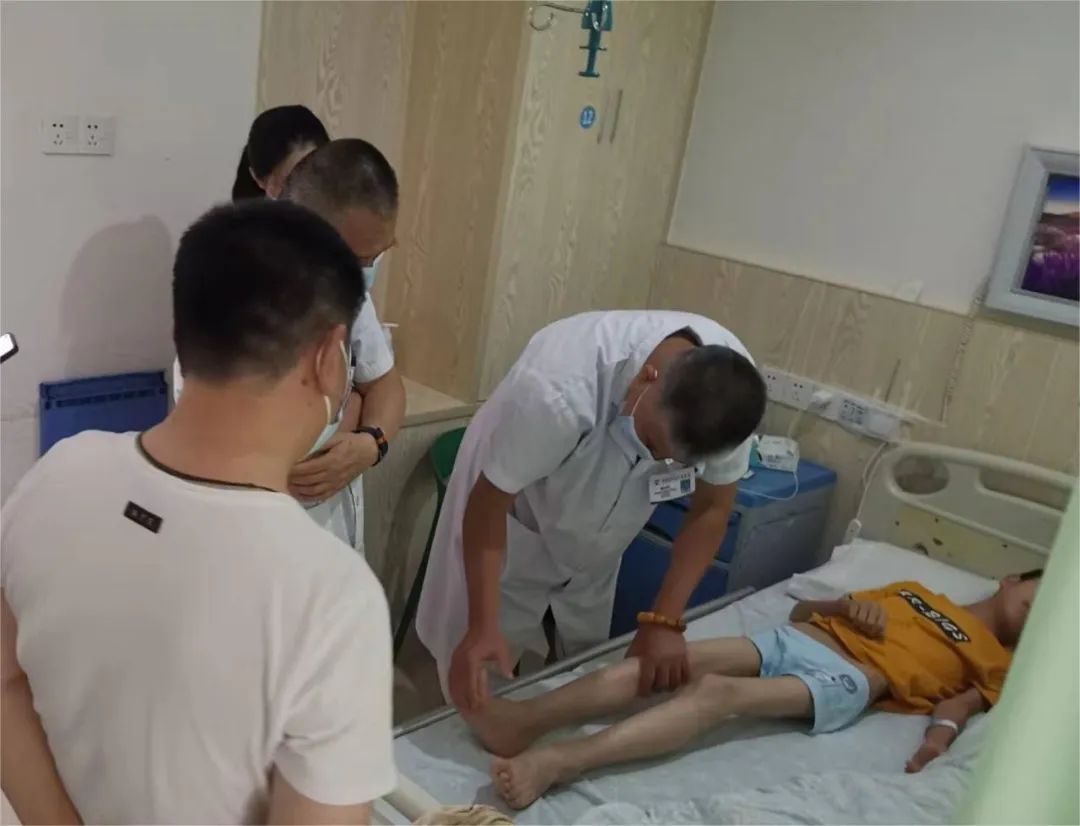
7月13日,冬冬在唐华利副主任医师的的操作下顺利完成了第一次诺西那生钠的鞘内注射术,过程顺利,术后无不适。冬冬已经于7月14日顺利出院,并计划在14天后再次返院接受第二次负荷剂量治疗。
王菲主任介绍,桂林市妇幼保健院设有儿童神经遗传专科,且是目前桂北地区开展注射诺西那生钠治疗的首家医疗机构。桂北地区确诊的SMA的患儿目前已在我院儿科集中登记,之后将陆续纳入药物治疗和多学科管理的轨道。
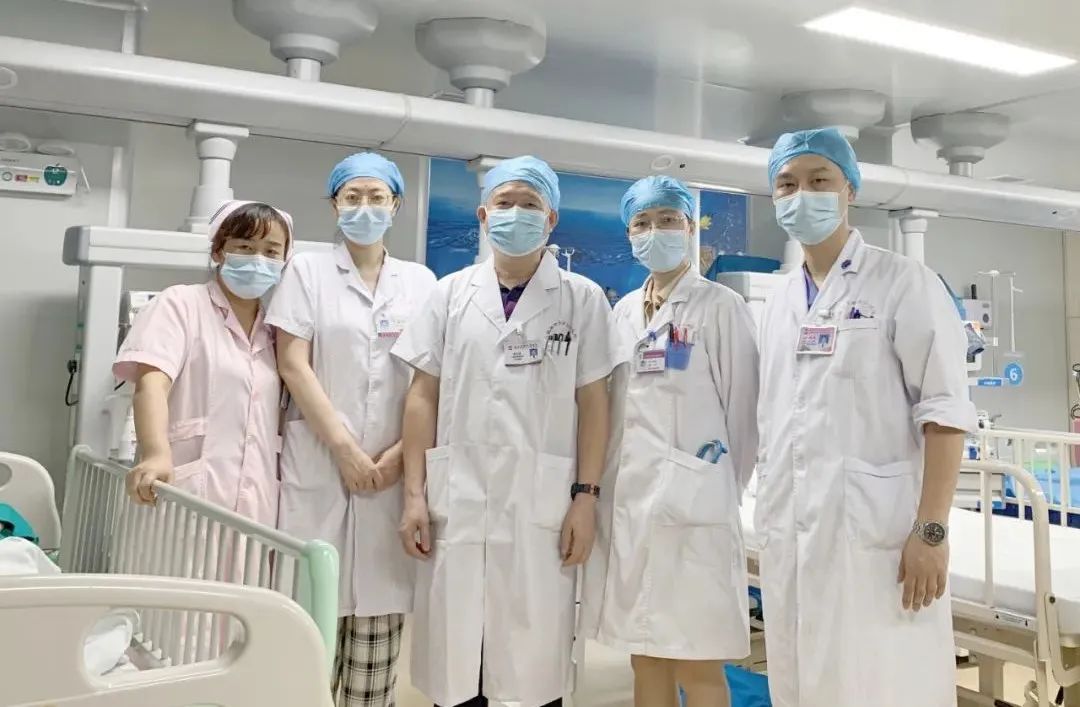
卢浩华院长表示,医改的核心要义是一切为了人民的健康,医院此举不仅是医院推动党史学习教育走深走实,纵深推进“清廉医风便民惠民”和“我为群众办实事”活动的体现,也是医院履行职责、贯彻落实医疗改革和改善医疗服务水平的重要举措。医院将继续在承担国家和广西妇幼健康惠民服务项目实施的基础上,不断的提升医疗质量和医疗服务水平,为大桂林及周边地区妇女儿童提供全生命周期全方位的医疗保健服务。
科普多一点:
基因筛查是预防的第一步
在普通人群中,每40-50个人中就有一个携带SMA致病基因,如果两个SMA致病基因的携带者婚育,他们的后代有25%的可能患上SMA。
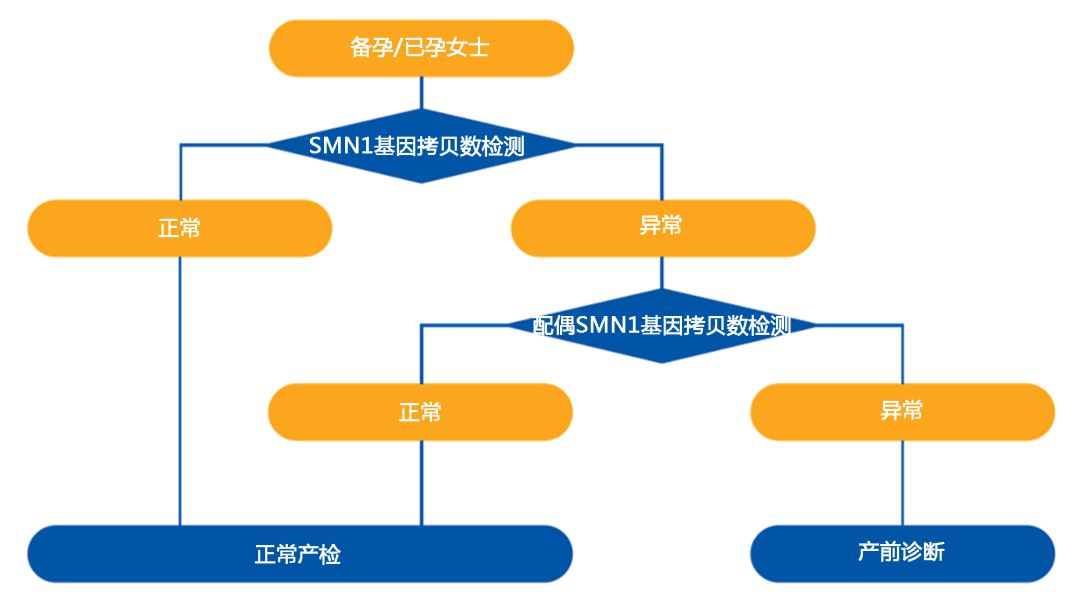
我国人群SMA基因的携带频率高,因此,通过携带者筛查,孕前检测,产前诊断及辅助生殖技术,可以明显降低SMA患儿的出生率,提高人口质量,减轻社会负担。美国妇产科学院推荐所有准备怀孕或已经怀孕的女性以及有家族成员诊断SMA的所有人进行SMA携带者筛查,预防SMA,产前基因诊断是关键。

桂林市妇幼保健院专家建议,夫妻双方在孕前或孕期筛查是否是SMA携带者。如果夫妻双方都是携带者,在孕前可以通过第三代试管婴儿避免生育SMA患儿。在孕期,孕妇则需要经过绒毛穿刺或羊膜腔穿刺,对胎儿进一步产前诊断。
延伸阅读
SMA是一种常染色体隐性遗传神经变性病,其特征是脊髓前角运动神经元及延髓LMN的变性,引起进行性肌肉无力和萎缩,属于典型MND的一种。
已有越来越多的基因突变被报道与SMA表型相关,它们在遗传学上和临床上具有明显异质性。最常见的形式由染色体5q13上SMN1基因的等位基因缺失或突变引起,故又被称为SMN1-连锁SMA。
分子遗传学可以通过检测SMN1基因外显子7的纯合缺失来证实SMA的诊断。SMN蛋白及表型差异与位于SMN1基因附近的修饰基因SMN2有关,虽然SMN1的丧失对SMA的发病机制至关重要,但疾病的严重程度与SMN2的拷贝数有关。
SMA表型根据发病年龄和临床过程分为0~4型:
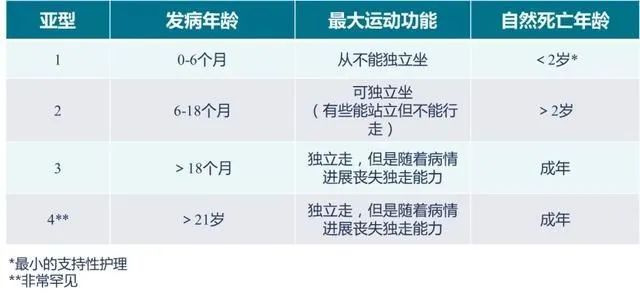
SMA 0型为产前发作,出生时出现严重无力,肌张力低,反射消失。通常1~6个月即因呼吸衰竭而死亡。
SMA 1型(婴儿脊髓性肌萎缩症或Werdnig-Hoffmann病)通常在出生后6个月之前出现。症状进展迅速,大多数婴儿在2岁前死于呼吸衰竭。
SMA 2型(中间型)和SMA 3型(Kugelberg-Welander病)病情相对较轻。SMA 2型出现于3~15个月,
SMA 3型通常18个月到成年起病,并且进展为慢性病程。
SMA 4型于成人起病,是SMA最轻微的形式。所有形式的SMA患者都具有弥漫性对称的近端肌无力,下肢较上肢更明显,伴深反射减低或消失。
SMA的鉴别诊断范围广泛,根据发病年龄而有所不同。成人发病患者无明显UMN受累体征,病程进展缓慢,预后好。基因检测、肌电图运动单位数指数可显示出不同于典型ALS的特征。SMA是由SMN突变引起的单基因疾病,而ALS涉及多个基因以及环境因素。另一方面,蛋白组学和生物资料分析显示,ALS与SMA有共同的分子通路,提示不同类型的MND存在潜在的共同机制。

 返回顶部
返回顶部电话:0773 - 2884335(预约)
桂林市妇幼保健院(桂林市妇女儿童医院) Q Q:3075624875
凤北路院区地址:广西桂林市叠彩区凤北路20号
东镇路院区地址:广西桂林市叠彩区中山北路27号
女性健康管理中心地址:凤北路南院区
桂公网安备 45030302000197号 桂ICP备15006704号
桂林市妇幼保健院(桂林市妇女儿童医院) 版权所有 技术支持 :云腾信息









